Today, Carlos fulfilled all requirements to the award of the Ph.D. degree….
Congratulations Dr. Carlos!!!! 🙂
Today, Carlos fulfilled all requirements to the award of the Ph.D. degree….
Congratulations Dr. Carlos!!!! 🙂
Carlos Eduardo Sequeiros Borja and Bartłomiej Surpeta will go last leg of their doctoral journey in our laboratory this October.
Best of luck!
https://biologia.amu.edu.pl/wydarzenia/obrony-rozpraw-doktorskich/publiczna-obrona-rozprawy-doktorskiej-mgr.-carlosa-eduardo-sequeirosa-borja
https://biologia.amu.edu.pl/wydarzenia/obrony-rozpraw-doktorskich/publiczna-obrona-rozprawy-doktorskiej-mgr.-bartlomieja-surpety
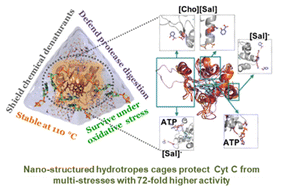
Bharadwaj P,* Sarkar DK,* Bisht M, Shet SM, Nataraj SK, Lokesh V, Franklin G,# Brezovsky J,# Mondal D,# 2023: Nano-structured hydrotrope-caged cytochrome c with boosted stability in harsh environments: a molecular insight. Green Chemistry 25: 6666-6676. full text dataset
Green and nano-structured catalytic media are vital for biocatalysis to attenuate the denaturation tendency of biocatalysts under severe reaction conditions. Hydrotropes with multi-faceted physiochemical properties represent promising systems for sustainable protein packaging. Herein, the ability of adenosine-5′-triphosphate (ATP) and cholinium salicylate ([Cho][Sal]) ionic liquid (IL) to form nano-structures and to nano-confine Cytochrome c (Cyt c) enhanced the stability and activity under multiple stresses. Experimental and computational analyses were undertaken to explain the nano-structured phenomenon of ATP and IL, structural organizations of nano-confined Cyt c, and site-specific interactions that stabilize the protein structure. Both ATP and IL form nano-structures in aqueous media and could cage Cyt c via multiple nonspecific soft interactions. Remarkably, the engineered molecular nano-cages of ATP (5–10 mM), IL (300 mg mL−1), and ATP + IL surrounding Cyt c resulted in 9-to-72-fold higher peroxidase activity than native Cyt c with exceptionally high thermal tolerance (110 °C). The polar interactions with the cardiolipin binding site of Cyt c, mediated by hydrotropes, were well correlated with the increased peroxidase activity. Furthermore, higher activity trends were observed in the presence of urea, GuHCl, and trypsin without any protein degradation. Specific binding of hydrotropes in highly mobile regions of Cyt c (Ω 40–54 residues) and enhanced H-bonding with Lys and Arg offered excellent stability under extreme conditions. Additionally, ATP effectively counteracted reactive oxygen species (ROS)-induced denaturation of Cyt c, which was enhanced by the [Sal] counterpart of IL. Overall, this study explored the robustness of nano-structured hydrotropes to have a higher potential for protein packaging with improved stability and activity under extreme conditions. Thus, the present work highlights a novel strategy for real-time industrial biocatalysis to protect mitochondrial cells from ROS-instigated apoptosis.
Pakuła K,* Sequeiros-Borja C,* Biała-Leonhard W,* Pawela A, Banasiak J, Bailly A, Radom M, Geisler M, Brezovsky J,# Jasiński M,# 2023: Restriction of access to the central cavity is a major contributor to substrate selectivity in plant ABCG transporters. Cellular and Molecular Life Sciences 80: 105. full text

ABCG46 of the legume Medicago truncatula is an ABC-type transporter responsible for highly selective translocation of the phenylpropanoids, 4-coumarate, and liquiritigenin, over the plasma membrane. To investigate molecular determinants of the observed substrate selectivity, we applied a combination of phylogenetic and biochemical analyses, AlphaFold2 structure prediction, molecular dynamics simulations, and mutagenesis. We discovered an unusually narrow transient access path to the central cavity of MtABCG46 that constitutes an initial filter responsible for the selective translocation of phenylpropanoids through a lipid bilayer. Furthermore, we identified remote residue F562 as pivotal for maintaining the stability of this filter. The determination of individual amino acids that impact the selective transport of specialized metabolites may provide new opportunities associated with ABCGs being of interest, in many biological scenarios.
Sequeiros-Borja C, Surpeta B, Marchlewski I, Brezovsky J, 2022: Divide-and-conquer approach to study protein tunnels in long molecular dynamics simulations. MethodsX (in press – DOI: 10.1016/j.mex.2022.101968). full text

Nowadays, molecular dynamics (MD) simulations of proteins with hundreds of thousands of snapshots are commonly produced using modern GPUs. However, due to the abundance of data, analyzing transport tunnels present in the internal voids of these molecules, in all generated snapshots, has become challenging. Here, we propose to combine the usage of CAVER3, the most popular tool for tunnel calculation, and the TransportTools Python3 library into a divide-and-conquer approach to speed up tunnel calculation and reduce the hardware resources required to analyze long MD simulations in detail. By slicing an MD trajectory into smaller pieces and performing a tunnel analysis on these pieces by CAVER3, the runtime and resources are considerably reduced. Next, the TransportTools library merges the smaller pieces and gives an overall view of the tunnel network for the complete trajectory without quality loss.