Bharadwaj P,* Sarkar DK,* Bisht M, Shet SM, Nataraj SK, Lokesh V, Franklin G,# Brezovsky J,# Mondal D,# 2023: Nano-structured hydrotrope-caged cytochrome c with boosted stability in harsh environments: a molecular insight. Green Chemistry 25: 6666-6676. full text dataset
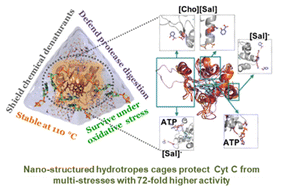
Green and nano-structured catalytic media are vital for biocatalysis to attenuate the denaturation tendency of biocatalysts under severe reaction conditions. Hydrotropes with multi-faceted physiochemical properties represent promising systems for sustainable protein packaging. Herein, the ability of adenosine-5′-triphosphate (ATP) and cholinium salicylate ([Cho][Sal]) ionic liquid (IL) to form nano-structures and to nano-confine Cytochrome c (Cyt c) enhanced the stability and activity under multiple stresses. Experimental and computational analyses were undertaken to explain the nano-structured phenomenon of ATP and IL, structural organizations of nano-confined Cyt c, and site-specific interactions that stabilize the protein structure. Both ATP and IL form nano-structures in aqueous media and could cage Cyt c via multiple nonspecific soft interactions. Remarkably, the engineered molecular nano-cages of ATP (5–10 mM), IL (300 mg mL−1), and ATP + IL surrounding Cyt c resulted in 9-to-72-fold higher peroxidase activity than native Cyt c with exceptionally high thermal tolerance (110 °C). The polar interactions with the cardiolipin binding site of Cyt c, mediated by hydrotropes, were well correlated with the increased peroxidase activity. Furthermore, higher activity trends were observed in the presence of urea, GuHCl, and trypsin without any protein degradation. Specific binding of hydrotropes in highly mobile regions of Cyt c (Ω 40–54 residues) and enhanced H-bonding with Lys and Arg offered excellent stability under extreme conditions. Additionally, ATP effectively counteracted reactive oxygen species (ROS)-induced denaturation of Cyt c, which was enhanced by the [Sal] counterpart of IL. Overall, this study explored the robustness of nano-structured hydrotropes to have a higher potential for protein packaging with improved stability and activity under extreme conditions. Thus, the present work highlights a novel strategy for real-time industrial biocatalysis to protect mitochondrial cells from ROS-instigated apoptosis.


